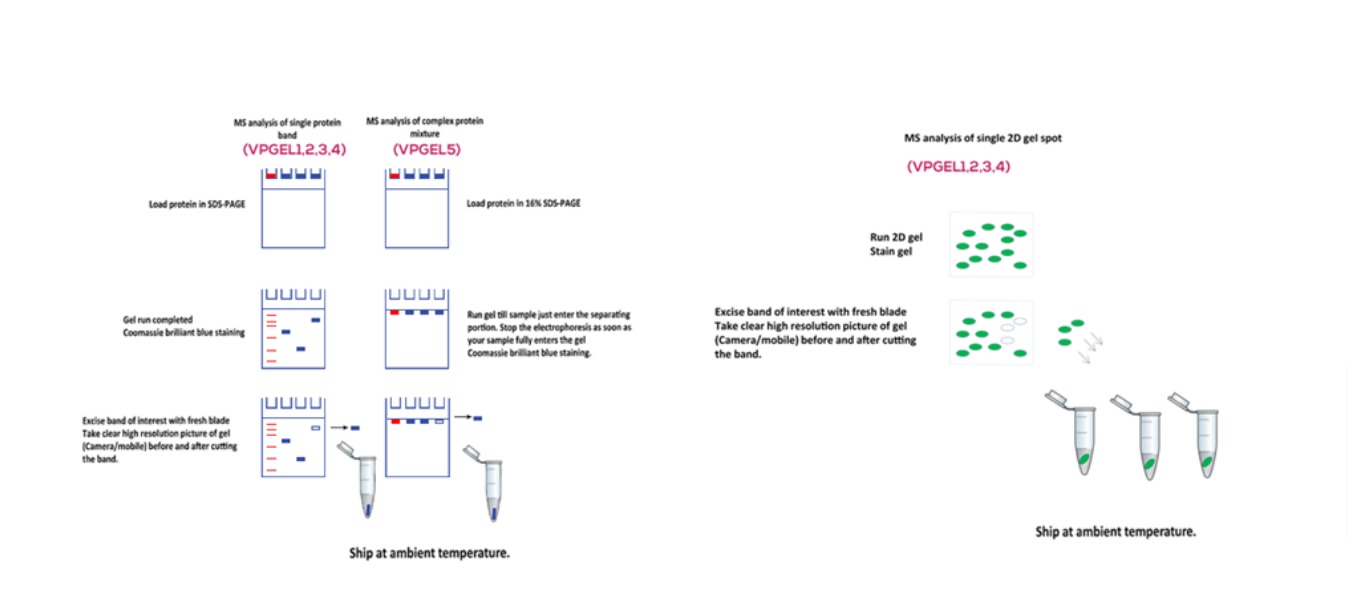
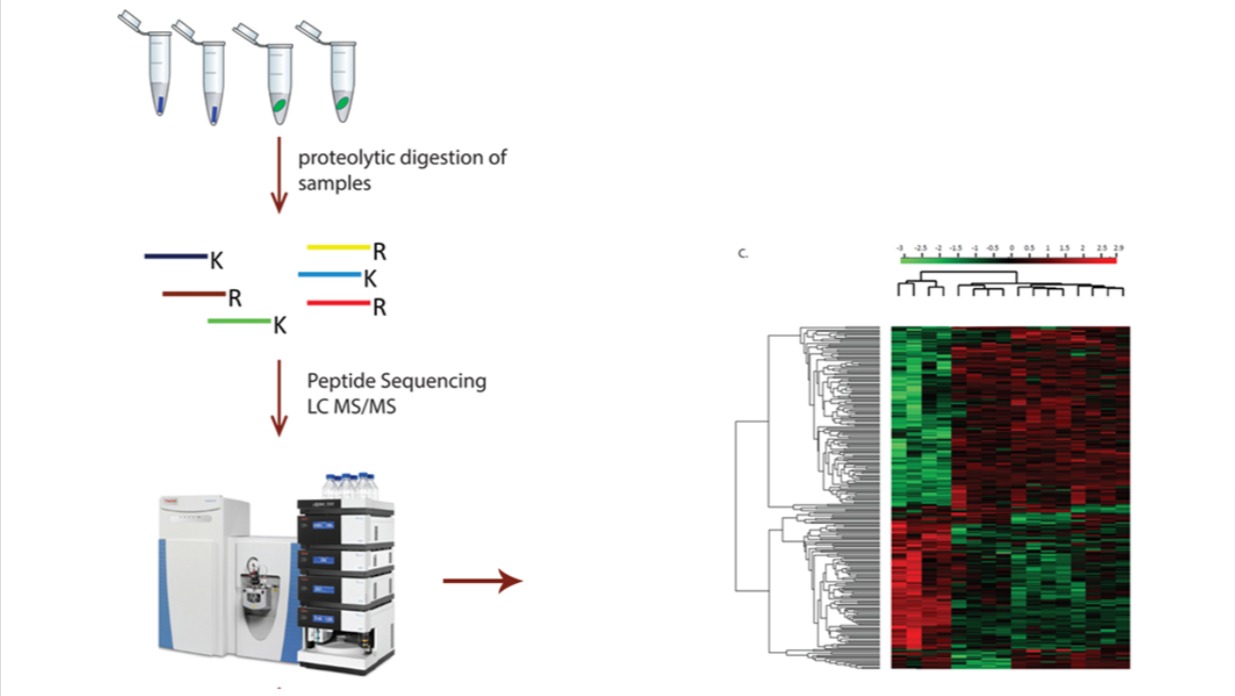
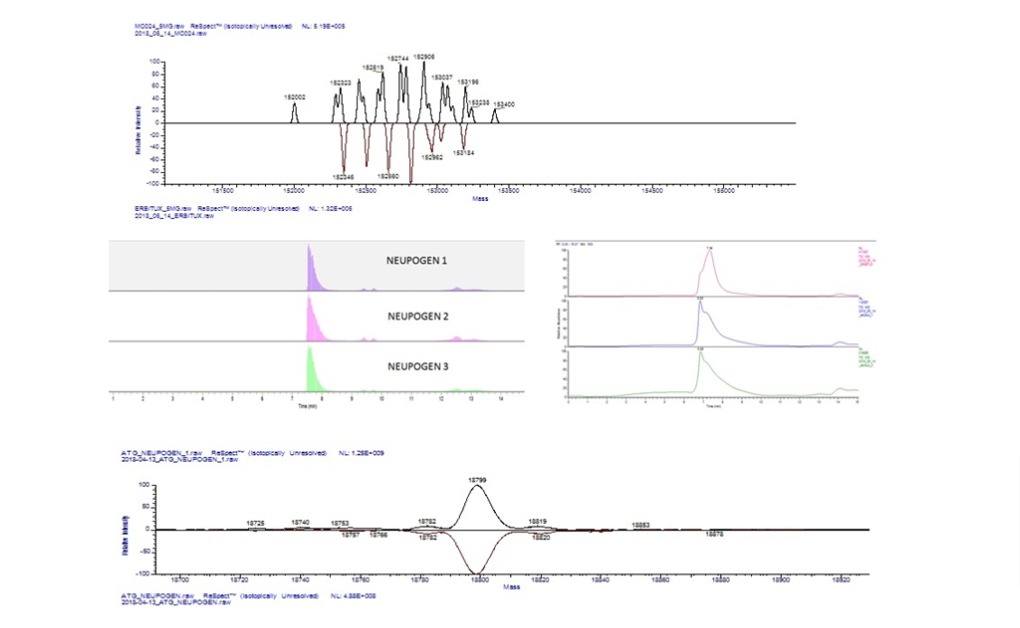
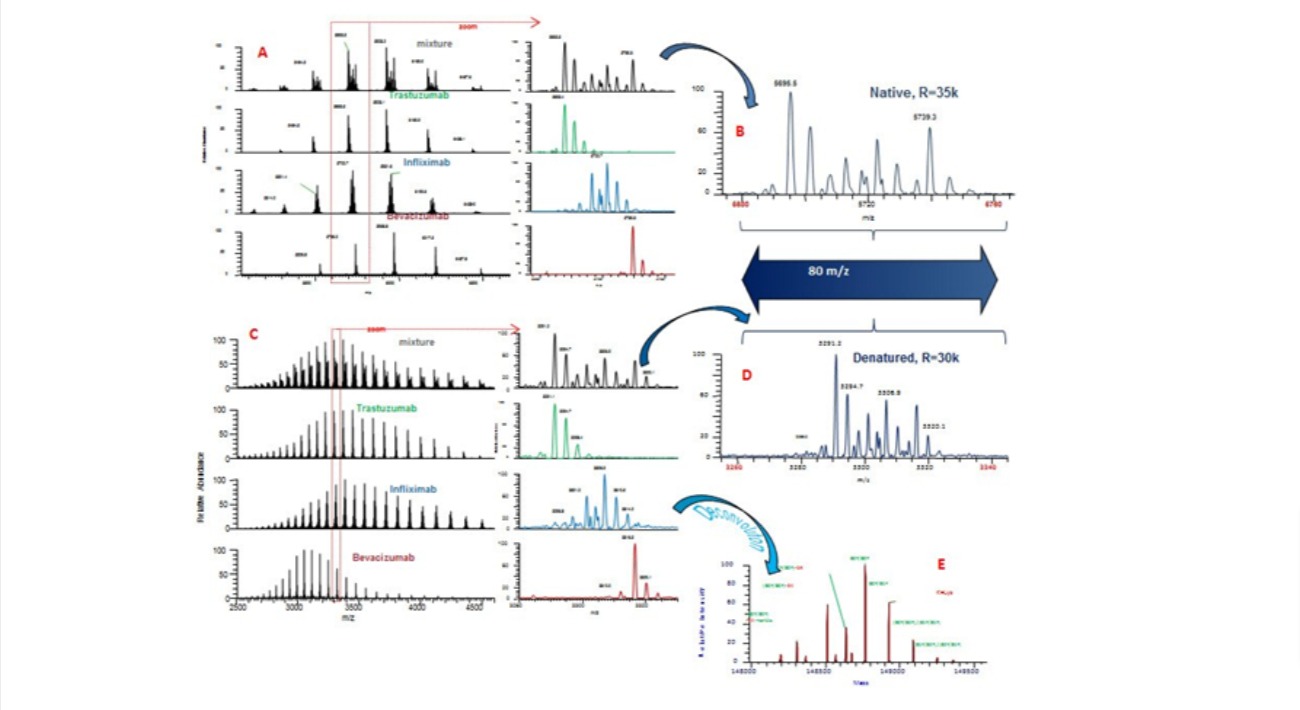
Mass spectrometry (MS) is widely used in the characterization of biomolecules including
peptide and protein therapeutics. These biotechnology products have seen rapid growth over
the past few decades and continue to dominate the global pharmaceutical market. Advances in
MS instrumentation and techniques have enhanced protein characterization capabilities and
supported an increased development of biopharmaceutical products.
Intact Mass Analysis
Intact mass analysis is the assessment of a protein’s total molecular
weight by mass spectrometry (MS) without prior digestion or fragmentation of the molecule of
interest. Molecular weight determination forms part of the ICH Q6B guidelines for
physicochemical analysis of biological products: what use is this information in the
analytical characterization of biopharmaceuticals?
High resolution MS allows the average molecular weight of large
proteins, for example monoclonal antibodies (mAbs), to be measured with accuracy better than
± 0.01%. The observed mass can then be compared to the expected mass for a given amino acid
sequence.
Peptide Mapping
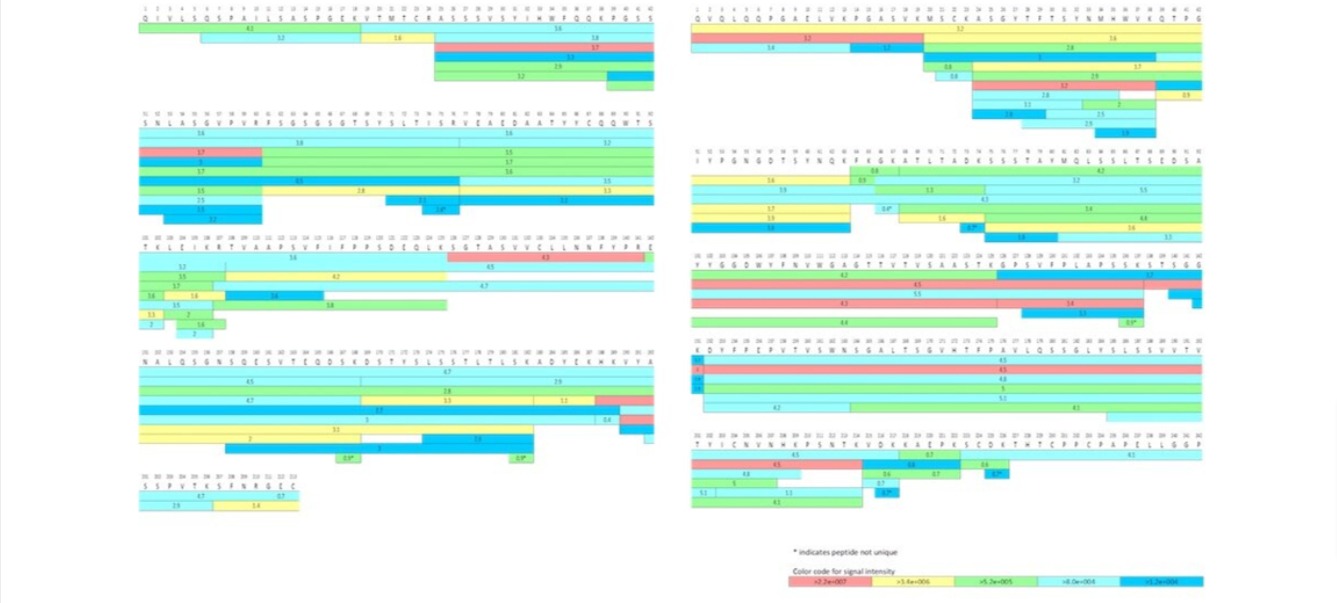
Peptide mapping is a critical workflow in
biotherapeutic protein characterization and is essential for elucidating the primary amino
acid structure of proteins. For recombinant protein pharmaceuticals, such as monoclonal
antibodies (mAbs) and antibody-drug conjugates (ADCs), peptide mapping is used for proof of
identity, primary structural characterization and quality assurance/quality control (QA/QC).
Global regulatory agencies, including US Food and Drug Administration (US FDA) and European
Medicines Agency (EMA), look to harmonized guidelines from the International Council for
Harmonisation (ICH). ICH Q6B covers the test procedures and acceptance criteria for biologic
drug products, and specifies the use of peptide mapping as a critical quality test procedure
for drug characterization used to confirm desired product structure for lot release
purposes.
In order to generate a peptide map, the therapeutic protein must first be digested into its
constituent peptides via a chemical or enzymatic reaction. Robust separation and
identification of the resultant peptides then provides insight into a protein’s full
sequence information; displaying each amino acid component and the surrounding amino acid
microenvironment, including disulfide linkage information. Structural characterization at
this level highlights post translational modifications (PTMs) such as site-specific
glycosylation, amino acid substitutions (sequence variants) and/or truncations which may
result from erroneous transcription of complementary DNA. Within a bioproduction
environment, peptide mapping is necessary for manufacturing process monitoring and QC. It
facilitates product comparability testing, which is necessary to identify any
product-related impurities, such as deamidation and/or oxidation following any formulation,
manufacturing process or storage change.
Due to its complexity and inherent variability, peptide mapping is generally performed in a
comparative manner; for example, biosimilars would be compared to a reference or control
substance, such as the innovator biologic, in a side-by-side experiment. An in-depth
analysis is then required to identify minor and even isobaric differences in protein primary
structure.
N-terminal and C- terminal sequencing
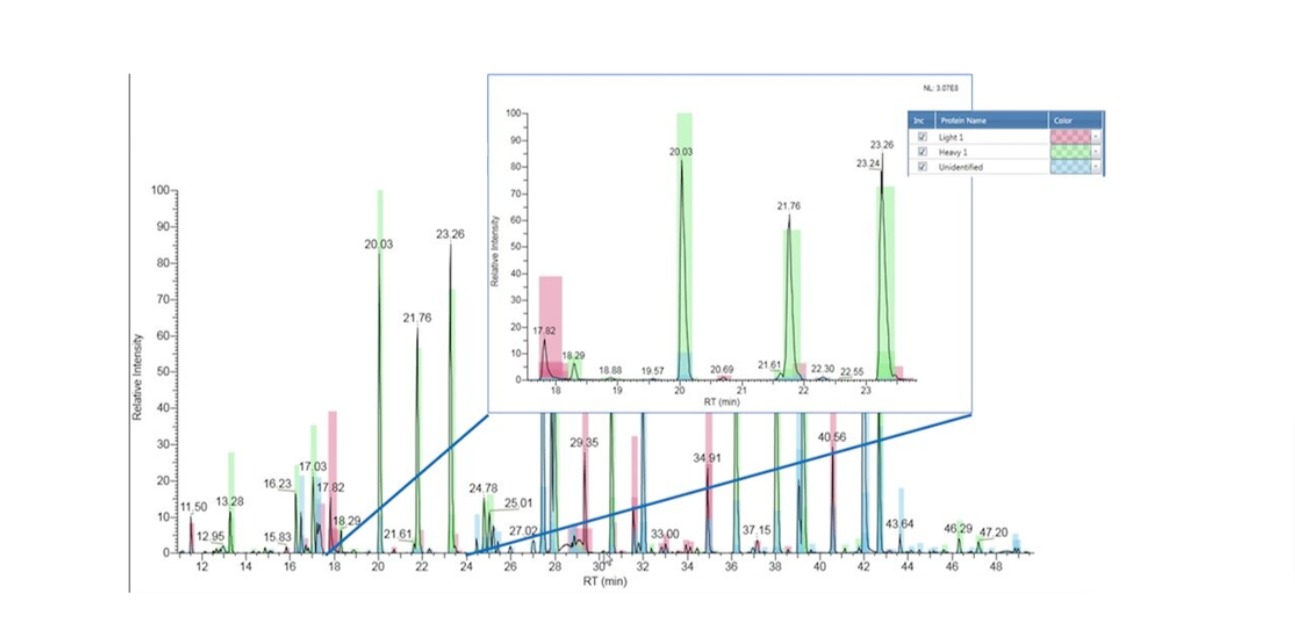
N-terminal sequencing is widely accepted as a reliable tool for protein
characterization throughout all stages of drug discovery and biopharmaceutical
manufacturing. The two major direct methods of protein N-terminal sequencing are Edman
degradation and mass spectrometry. A strategic approach to the enzyme digestion via an in
silico digest selectively optimises the N-terminal peptide fragmentation to achieve suitable
peptides of 20-50 amino acid residues.The intact peptide is then deliberately fragmented
within a mass spectrometer in order to gain structural information from the fragment ions
Due to its complexity and inherent variability, peptide mapping is generally performed in a
comparative manner; for example, biosimilars would be compared to a reference or control
substance, such as the innovator biologic, in a side-by-side experiment. An in-depth
analysis is then required to identify minor and even isobaric differences in protein primary
structure.